
脂肪伯胺是天然产物和药物分子中广泛存在的关键结构单元,也是有机合成中的重要砌块。因此,脂肪伯胺的高效合成和后修饰一直是研究的热点。然而,相较于二级、三级脂肪胺,脂肪伯胺因易形成稳定的双(胺)金属配合物,或发生α-氧化、Buchwald-Hartwig偶联等副反应,其后期官能团化面临着更大挑战。
氧化环化是构建重要的碳环和杂环体系,尤其是多环体系的高效策略之一。近年来,基于脂肪伯胺α-C(sp3)-H官能团化的氧化环化反应已成功实现了咪唑,三唑,苯并恶唑,噻唑,苯并噻唑和γ-内酰胺等多种重要氮杂环的高效构建。然而,相对于活化的α-C(sp3)-H键,实现更具挑战性的β-C(sp³)-H键官能团化,并以此构建结构多样的含氮杂环,仍是该领域亟待解决的关键科学问题。
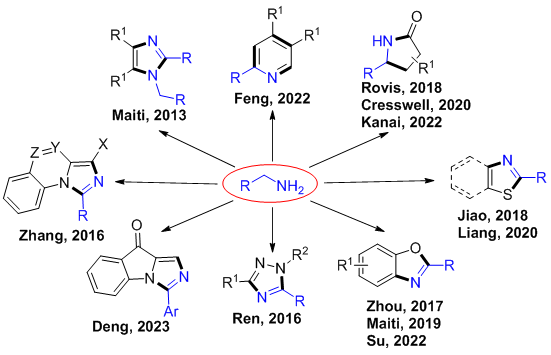
针对这一挑战,南华大学药学院陈劲进博士/王震教授团队近期取得了系统性研究进展。团队开发了一种碘促进的,链状脂肪伯胺、芳香醛和碘化铵四组分反应,高效构建多取代嘧啶类化合物的新方法。该反应创新性地以脂肪伯胺作为C-C-N合成子,芳香醛作为C1合成子,碘化铵作为环境友好的氮源,在“一锅”中实现了脂肪伯胺的α-C(sp3)-H和β-C(sp3)-H键官能化。该方法为具有重要生物活性的多取代嘧啶类化合物的合成提供了简洁、高效的新途径。相关成果发表于《Organic & Biomolecular Chemistry》(IF=2.7,中科院3区),论文第一作者为药学院硕士研究生刘康,通讯作者为药学院彭颖博士、王震教授和陈劲进博士。

在此基础上,团队进一步将该策略拓展至环状脂肪伯胺体系,在无金属条件下,利用环状脂肪伯胺、醛与碘化铵为原料,通过脂肪伯胺的氧化环化与β-C(sp³)-H官能团化反应,成功实现了多种半饱和稠合嘧啶类化合物的选择性合成。更重要的是,在I2/DMSO/O2氧化体系下,该反应可“一锅”直接转化为喹唑啉类衍生物,无需中间体的分离,显著提升了合成效率。该方法为重要的半饱和稠合嘧啶与喹唑啉类优势骨架提供了简单高效、环境友好的合成新方法。相关成果发表于《Organic Chemistry Frontiers》(中科院二区,IF=4.7),论文第一作者为药学院硕士研究生刘康,通讯作者为湘潭大学化学学院邓国军教授、南华大学药学院王震教授和陈劲进博士,南华大学药学院为第一通讯单位。
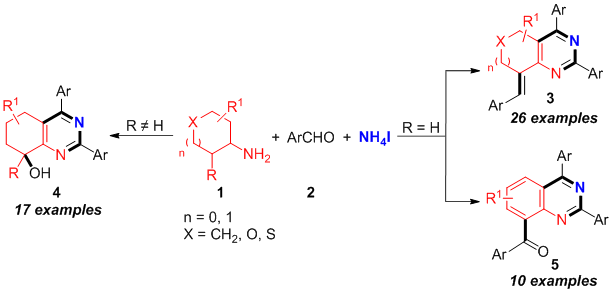
该系列工作突破了脂肪伯胺后修饰的传统思路,通过发展新颖的脂肪伯胺氧化环化策略,成功实现了其惰性β-C(sp³)-H键的选择性官能团化,为快速构建结构多样、复杂的含氮杂环化合物提供了简单高效的新方法,对相关药物先导化合物的发现与优化具有积极意义。
论文链接: